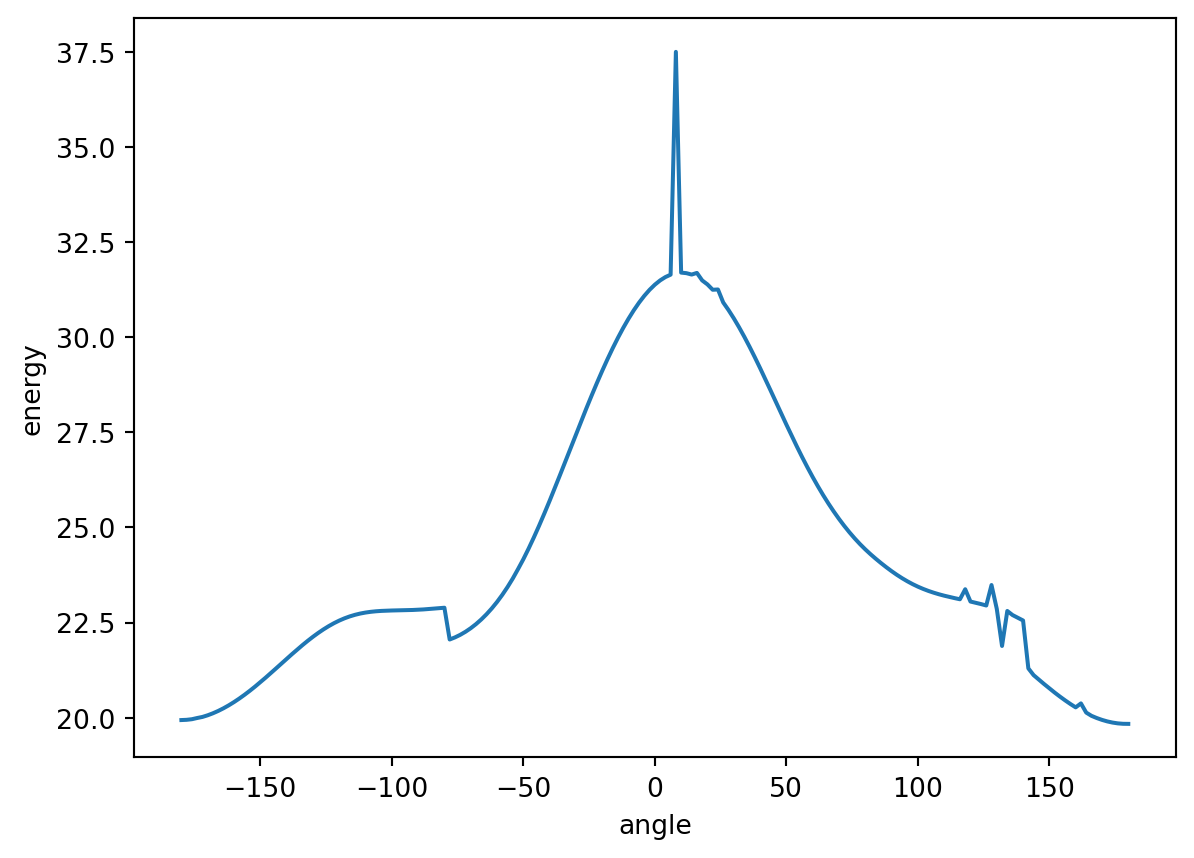
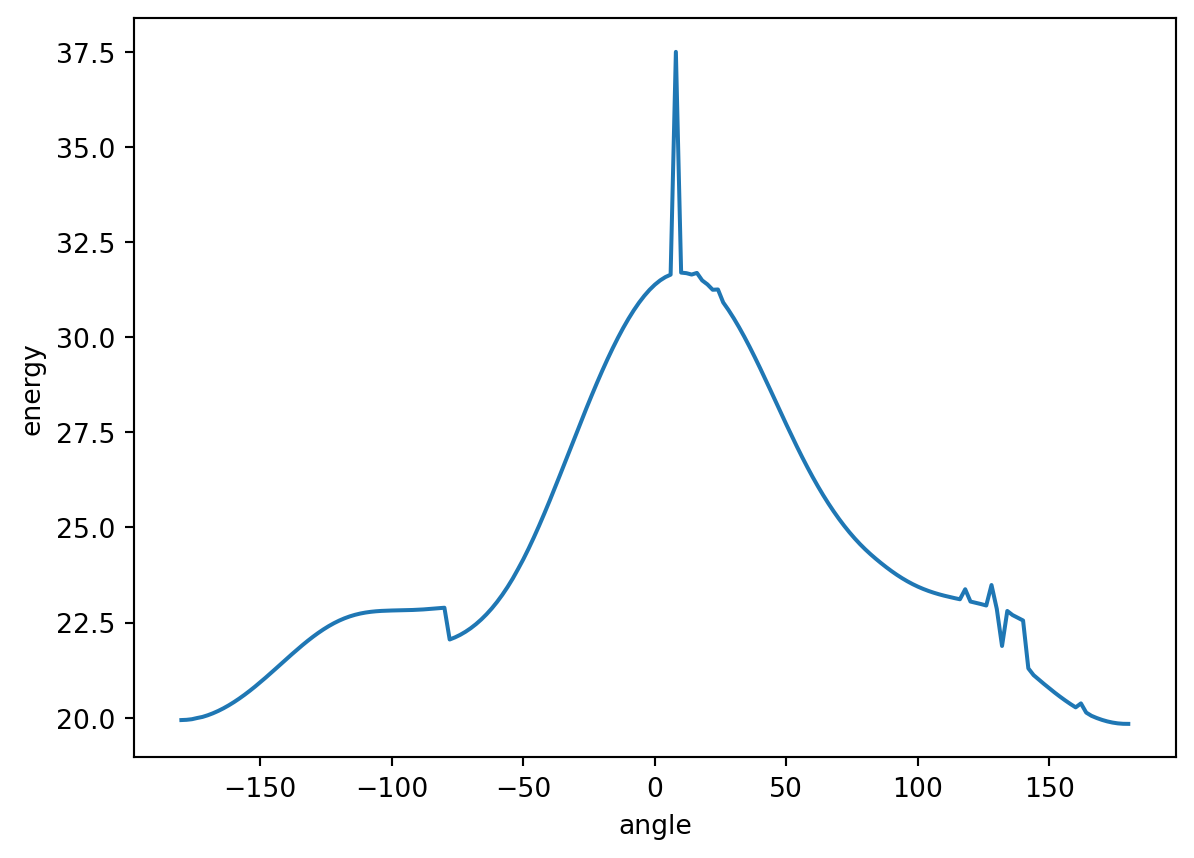
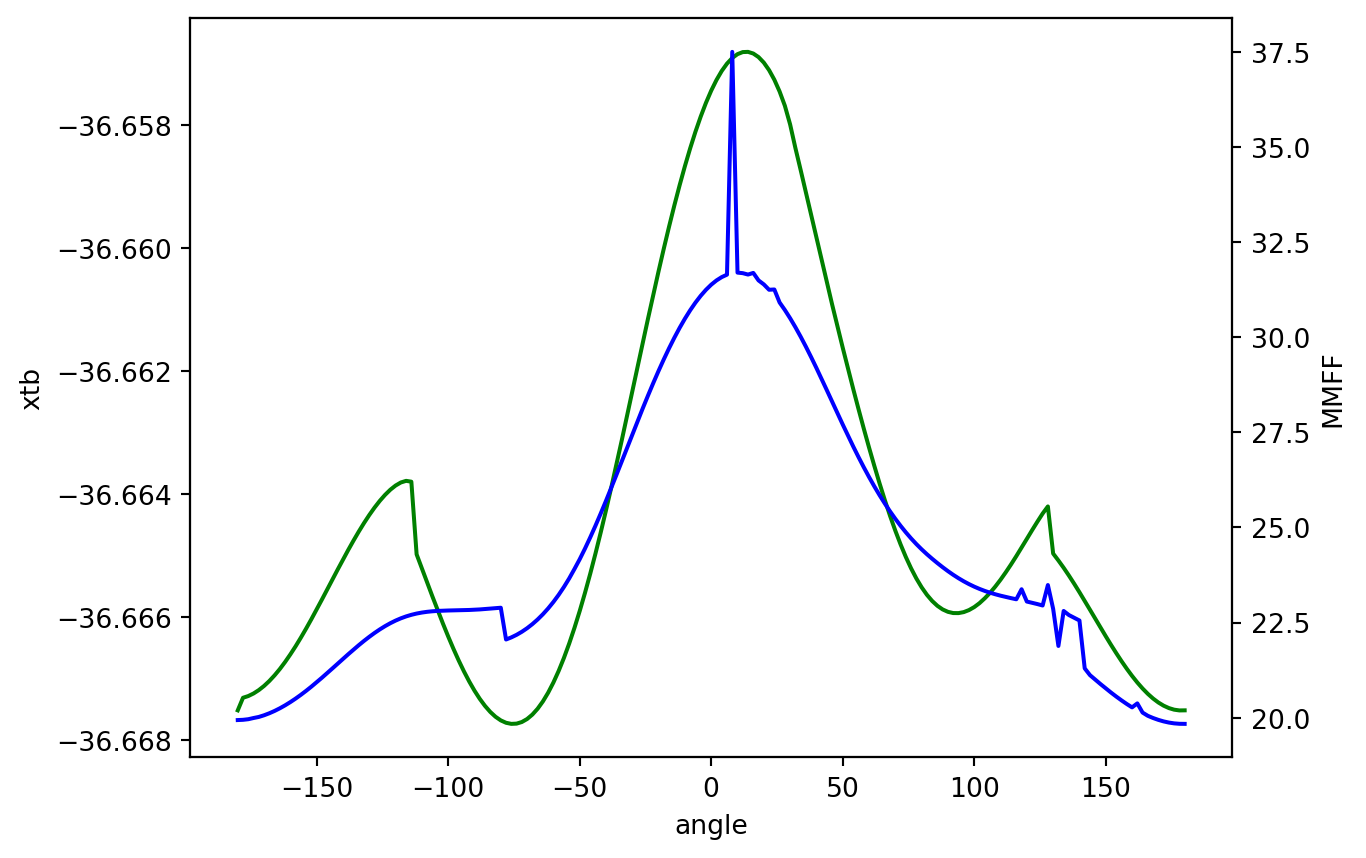
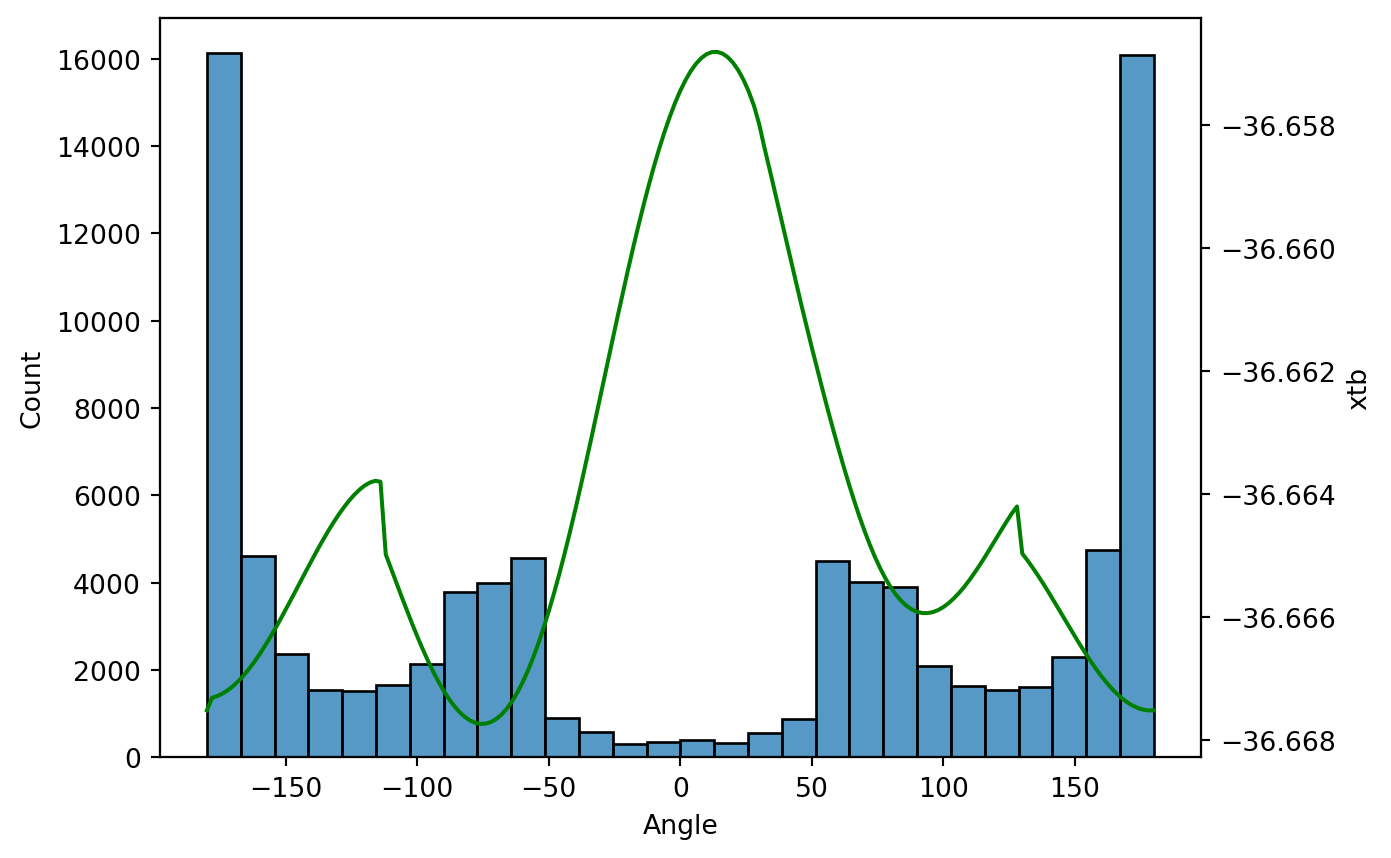
[-36.667513607102, -36.667309140177, -36.667281342484, -36.667239620489, -36.667184526773, -36.667117066126, -36.667037048311, -36.666945068211, -36.666842035773, -36.666727935772, -36.666604060224, -36.666472299839, -36.666331713852, -36.666184708292, -36.666031740213, -36.665874503518, -36.665713626478, -36.665550482059, -36.66538825599, -36.665225539683, -36.665065448331, -36.664908207787, -36.664756200554, -36.664609973167, -36.664471049714, -36.664340711715, -36.664220117532, -36.664110543681, -36.664013292087, -36.663929734669, -36.663861827436, -36.663811930617, -36.663785358953, -36.663798737338, -36.664979277748, -36.66520794, -36.665435059634, -36.665662744478, -36.665885124937, -36.666103090883, -36.66631293046, -36.666513718719, -36.666703584448, -36.666881133011, -36.66704466986, -36.667193605558, -36.667326090946, -36.667441655993, -36.667539140451, -36.667617824736, -36.667676838596, -36.667715513415, -36.667733191444, -36.667729259909, -36.667703174391, -36.667654498956, -36.667582681905, -36.667487621575, -36.667369122472, -36.667227133502, -36.667061760273, -36.666873378488, -36.666662375664, -36.666429356721, -36.666175209343, -36.665900780799, -36.665607020687, -36.665295092731, -36.664966346322, -36.664622108195, -36.664264015668, -36.663893894368, -36.663513617705, -36.663125457688, -36.662731505843, -36.662334108898, -36.661935609452, -36.661538373839, -36.661144767473, -36.660757102954, -36.660377606895, -36.660008348515, -36.659651419864, -36.659308746758, -36.658982148344, -36.65867330741, -36.658383810707, -36.658115108876, -36.657868511125, -36.657644953523, -36.657446596701, -36.657273573907, -36.65712674186, -36.657007487457, -36.656916139656, -36.656853449108, -36.656820030572, -36.65681652035, -36.656843563867, -36.656901487252, -36.656991065908, -36.657113141754, -36.657268773622, -36.657460253667, -36.657691643264, -36.657995009871, -36.658377284791, -36.658734779587, -36.659101032723, -36.65947082494, -36.65983907601, -36.660205762865, -36.660568445789, -36.660925547559, -36.661276290406, -36.661622045455, -36.661961151255, -36.662293542142, -36.662618617756, -36.662935539654, -36.663243196539, -36.663539682536, -36.663825629787, -36.664097088781, -36.66435469442, -36.664595178208, -36.664818301433, -36.66502278909, -36.665207515844, -36.66537160691, -36.665514564786, -36.665636254032, -36.665736540925, -36.66581574069, -36.665874412542, -36.665913153582, -36.665933064251, -36.665933964303, -36.665917822483, -36.665884812253, -36.665836013797, -36.665772163003, -36.665693615985, -36.665604836221, -36.665503223322, -36.665391578063, -36.665270323778, -36.665142434253, -36.665007952167, -36.664869075563, -36.664728209023, -36.664586960773, -36.664449514058, -36.664316973593, -36.664201080863, -36.664966618261, -36.665079218636, -36.665198049586, -36.665324534034, -36.665457378424, -36.665594877696, -36.665735947942, -36.665879336208, -36.666023480717, -36.666167690138, -36.666309918055, -36.666449518595, -36.666584866403, -36.666714785838, -36.666838170524, -36.666953893713, -36.667061007153, -36.667158583164, -36.667245753906, -36.667321770371, -36.667385989984, -36.667437794223, -36.667476728724, -36.667502584037, -36.667514883009, -36.667513577254]
range(-180, 182, 2)
0 19.936138
1 19.942480
2 19.958324
3 19.989942
4 20.018098
...
176 19.900734
177 19.870128
178 19.849113
179 19.837853
180 19.835944
Name: energy, Length: 181, dtype: float64
{kind=link}